
Dr. Bánhegyi Róbert János Ph.D.
Metabolikus és molekuláris összefüggések
a rosszindulatú daganatok és a cukorbetegség között
ÖSSZEFOGLALÁS
Az utóbbi évtizedek orvosi kutatásainak egyik tartósan kiemelt területe a szénhidrátháztartás zavarai és a rosszindulatú daganatok közti metabolikus és molekuláris kapcsolatok részletes elemzése, amely folyamatosan újabb és újabb daganatellenes terápiák kifejlesztésének és bevezetésének lehetőségét vetíti előre. Epidemiológiai, preklinikai és klinikai vizsgálatok alapján ma már biztosan tudjuk, hogy az előrehaladott cukorbetegség számos daganat képződésében önálló rizikótényezőként szerepelhet, sőt a tumorkockázat növekedése akár már prediabéteszes kórállapotokban is jelentkezhet. Napjainkban ugyanakkor azt is teljes bizonyossággal kimondhatjuk, hogy a kétfajta kórkép közti kapcsolat fordított irányban is fennáll. Közismert tény, hogy a malignus daganatok a gazdaszervezetben olyan metabolikus és molekuláris változásokat idéznek elő, amelyek egy idő után a korai diabéteszre jellemző inzulinrezisztens állapotot eredményeznek. Ez a tumor által generált inzulinrezisztencia a betegek egy részénél akár másodlagos cukorbetegség kialakulásához is elvezethet, amit a jelenleg alkalmazott antineoplasztikus terápiák esetleges diabetogén effektusa csak tovább súlyosbíthat. Az utóbbi években a rosszindulatú daganatos megbetegedések és a 2-es típusú diabetes mellitus szoros kapcsolatának molekuláris okait feltárni kívánó kutatások a figyelmet a RAS és a PI3K jelátviteli utak központi szerepére irányították. Ezek megváltozott működése ugyanis a teljes sejtciklust, a komplett celluláris metabolizmust, a sejtek növekedését és proliferációját, vagyis a sejtek túlélését jelentősen befolyásolja, részben a tumorképződés és tumorprogresszió, részben pedig az inzulinrezisztencia létrejötte felé tolja. Ma már tudjuk, hogy a háttérben húzódó molekuláris ok-okozati viszonyok az IGF-receptorok, a RAS és a PI3K jelátviteli útvonalak működésének és kötődési pontjainak ismerete nélkül aligha érthetők meg. Éppen ezért munkánkban a legfontosabb metabolikus összefüggések rövid ismertetését követően a molekuláris kapcsolatok részleteire fókuszálunk.
Kulcsszavak: tumorkockázat, inzulinrezisztencia, másodlagos cukorbetegség, IGF-receptorok, PI3K és RAS jelátviteli utak
ONCODIABETOLOGY I.
Metabolic and molecular relationships between cancer and diabetes
ABSTRACT
In the recent decades numerous studies have been investigated the metabolic and molecular links between carbohydrate metabolic disorders and cancer, raising potential anti-tumor therapies.
Based on epidemiological, preclinical, and clinical studies, now we know that advanced diabetes is a distinct risk factor of the development of many tumors, and even prediabetes may lead to increased risk of developing cancer. Nowadays we can also state that the relationship is also present vice-versa. It is a well-known fact that malignancies cause metabolic and molecular changes in the host over time resulting in an insulin-resistant state, characteristic of early diabetes. The tumor induced insulin resistance may lead to the development of secondary diabetes in some patients with cancer. Furthermore the diabetogenic effect of present anticancer therapies may worsen the metabolic condition.
In recent years, research exploring the molecular causes of the association between malignancies and type 2 diabetes mellitus has highlighted the central role of RAS and PI3K signaling pathways.
The altered function these pathways significantly effect cell cycle, cellular metabolism, cell growth and proliferation, thus modifying cell survival, leading to tumorigenesis and tumor progression and to insulin resistance as well. Without understanding the associations between IGF receptors, RAS and PI3K signaling pathways the underlying molecular mechanism can not be understood. Therefore here we focus on these molecular mechanisms after a brief description of the most important metabolic connections between cancer and diabetes.
Keywords: cancer risk, insulin resistance, secondary diabetes, IGF receptors, RAS and PI3K signaling pathways
RÖVIDÍTÉSEK
AGE: (advanced glycation end products) előrehaladott glikációs végtermékek; AKT/PKB: (v-akt murine thymoma viral oncogene homolog 1/protein kinase B) v-akt egér thymoma virális onkogén homológ 1/protein-kináz B; AML: (acute myeloid leukemia) akut myeloid leukémia; AMP: (adenosine monophosphate) adenozin-monofoszfát; AMPK: (AMP-activated protein kinase) AMP-aktivált protein-kináz; ATP: (adenosine triphosphate) adenozin-trifoszfát; BAD: (BCL2-associated death promoter) BCL2-asszociált halálpromóter; BCL2: (B-cell leukemia/lymphoma 2 protein) B-sejtes leukémia/lymphoma 2 fehérje; CRC: (colorectal carcinoma) vastag- és végbélrák; DNS: (deoxyribonucleic acid) dezoxiribonukleinsav; EGF: (epidermal growth factor) epidermális növekedési faktor; ECOG PS: (Eastern Cooperative Oncology Group Performance Status); EGFR: (epidermal growth factor receptor) epidermális növekedési faktor receptor; ERK: (extracellular signal-regulated kinase) extracelluláris szignál által szabályozott kináz; FGF: (fibroblast growth factor) fibroblaszt növekedési faktor; FoxO: (forkhead box protein O); GAB1: (GRB2-associated binder) GRB2-asszociált kötőfehérje; GIST: (gastroinestinal stromal tumor) gasztrointesztinális stróma tumor; GLUT: (glucose transporter) glukóz transzporter; GRB: (growth factor receptor-bound protein) növekedési faktor receptorhoz kötött fehérje; HCC: (hepatocellular carcinoma) májsejtes rák; IFG: (impaired fasting glycaemia) kóros éhomi vércukorszint; IFN: (interferon); IGF: (insulin-like growth factor) inzulinszerű növekedési faktor; IGFBP: (IGF binding protein) IGF-kötő fehérje; IGFR: (IGF receptor) IGF-receptor; IGT: (impaired glucose tolerance) csökkent glukóztolerancia; IL: (interleukin); IR: (insulin resistance) inzulinrezisztencia; IRS: (insulin receptor substrate) inzulin receptor szubsztrát; LKB1: (liver kinase B1) májkináz B1; MAB: (monoclonal antibody) monoklonális antitest; MAPK: (mitogen-activated protein kinase) mitogén-aktivált protein-kináz; MDM: (murine double minute); MEK: (mitogen-activated protein kinase/extracellular signal-regulated kinase) mitogén-aktivált protein-kináz/extracelluláris szignál által szabályozott kináz; MTC: (medullary thyroid carcinoma) medulláris pajzsmirigyrák; mTOR: (mammalian target of rapamycin); mTORC: (mTOR Complex) mTOR Komplex; NET: (neuroendocrine tumor) neuroendokrin tumor; NFκB: (nuclear factor kappa B) nukleáris faktor kappa B; NHL: (non-Hodgkin lymphoma); NSCLC: (non-small cell lung cancer) nem-kissejtes tüdőrák; PDGF: (platelet-derived growth factor) vérlemezke eredetű növekedési faktor; PI3K: (phosphatidyl-inositol-3-kinase) foszfatidil-inozitol-3-kináz; PIF: (proteolysis-inducing factor) proteolízist indukáló faktor; PIK3CA: (PI3K catalytic subunit alpha) PI3K katalitikus alegysége alfa; PIK3R1: (PI3K regulatory subunit 1) PI3K reguláló alegysége 1; PTC: (papillary thyroid carcinoma) papilláris pajzsmirigyrák; PTEN: (tensin homolog deleted in chromosome 10) tenzin homológ elvesztése a 10. kromószómán; RAF: (rat sarcoma viral oncogene homolog B1) patkány szarkóma vírus onkogén homológ B1; RAS: (rat sarcoma viral oncogene homolog) patkány szarkóma vírus onkogén homológ; RCC: (renal cell carcinoma) vesesejtes rák; ROS: (reactive oxygen species) reaktív oxigén-származékok; RTK: (receptor tyrosine kinase) receptor tirozin-kináz; T1DM: (type 1 diabetes mellitus) 1-es típusú diabétesz mellitusz; T2DM: (type 2 diabetes mellitus) 2-es típusú diabétesz mellitusz; TGFβ: (transforming growth factor beta) transzformáló növekedési faktor béta; TKI: (tyrosine kinase inhibitor) tirozin-kináz inhibitor; TNFα: (tumor necrosis factor alpha) tumor nekrózis faktor alfa; TSC: (tuberous sclerosis complex) tuberózus szklerózis komplex; VEGF: (vascular endothelial growth factor) vaszkuláris endoteliális növekedési faktor; YAP: (yes-associated protein) igen-asszociált fehérje
BEVEZETÉS
Az utóbbi évtizedekben a szénhidrát-anyagcsere zavarainak - mind a prediabétesznek, vagyis a diabéteszt megelőző kezdeti kórállapotoknak (IR, IFG, IGT), mind pedig a manifeszt 2-es típusú cukorbetegségnek (T2DM) - a folyamatos térnyerése mellett általában a rosszindulatú daganatok gyakoriságának lassú és fokozatos emelkedését is tapasztalhatjuk. A kétféle megbetegedés populációs szintű megjelenésének szinkron változásait, egymással összefüggő terjedését epidemiológiai, obszervációs és mendeli randomizációs vizsgálatok egyaránt megerősítik [1]. Közös okaiknak és kórfolyamataiknak, a metabolizmusukra jellemző hasonlóságoknak és párhuzamosságoknak egyre részletesebb megismerése miatt a diabétesz és a daganatok képződése közti összefüggések napjainkban már alapvetően bizonyítottnak mondhatók [2, 3]. A gyakorlatban csupán a kapcsolat mértéke és mélysége, valamint a részleteinek minél teljesebb feltárása, majd az ezek alapján kijelölt támadáspontokat célzó gyógyszeres manipulációk kifejlesztése képezik a tudományos kutatások, viták célját és tárgyát [4].
A szénhidrát-anyagcsere zavarait kísérő fokozott tumorkockázat leginkább a cukorbetegség, elsősorban a T2DM kialakulásához vezető, illetve az ahhoz társuló inzulinrezisztencia létrejöttének, valamint a sejtoszlási folyamatok szabályozását és az immunrendszer működését is nagyban befolyásoló elhúzódó hyperglykaemiás állapotnak az eredménye [5]. A helyzet teljes megértését jelentősen nehezíti a kétfajta megbetegedés komplexitása is. Egyfelől a daganatos betegségnek rendkívül sokféle formája és típusa létezik a tumorok genetikai, hisztológiai, morfológiai jellemzői és változatos elsődleges szervi lokalizációi, valamint a beteg egyéni sajátosságai, neme, biológiai kora, általános állapota (ECOG PS), kardiális, renális, csontvelői és immunstátusza, továbbá egyéb szervi funkciói, társbetegségei, káros szenvedélyei, sőt intelligenciája és szociális helyzete szerint is. Másfelől a diabétesz sem egy egységesen leírható homogén betegség, hanem valójában egy olyan komplex kórállapot, amelyet a szénhidrát-anyagcsere eleve sokféle zavarán túl a zsír- és a fehérjeháztartás különböző típusú és mértékű kóros megváltozása is jellemez [6]. Ezt a bonyolult képet csak tovább árnyalja a cukorbetegség speciális altípusainak elkülönítése, amelyekben a különböző etiológiai tényezők mellett az eltérő patomechanizmusok is szerepet játszanak. Ezen tényezők és folyamatok mind részletesebb feltárása természetesen egyre inkább fokozza a diabétesz és a tumoros betegségek diagnosztikájának és kezelésének komplexitását, ám ezzel együtt a diabetológia és az onkológia területén is újszerű, a korábbiaktól sokszor hatékonyabb, innovatív terápiás lehetőségeket eredményez [7].
EPIDEMIOLÓGIAI, ETIOLÓGIAI ÖSSZEFÜGGÉSEK
Diabéteszhez társult tumor vagy daganathoz társult cukorbetegség?
A prediabétesz és a 2-es típusú cukorbetegség, valamint a malignus daganatok közötti összefüggést számos nagy metaanalízis következtetései is megerősítik [1, 2, 8]. Az IR, az IFG, az IGT és a T2DM fennállása - a teljes népesség körében tapasztalthoz képest - szignifikánsan nagyobb arányban fordul elő számos rákbetegség (pl. melanoma, NHL, HCC, RCC, CRC, hasnyálmirigy-, húgyhólyag-, prosztata-, emlő-, méhtest- és méhnyakrák stb.) esetén [9, 10, 11, 12, 13, 14, 15, 16, 17]. Az összefüggés fordítva is igaz, vagyis epidemiológiai és klinikai vizsgálatok eredményeivel alátámaszthatóan, az általános populációs adatokhoz viszonyítva a szénhidrátháztartás zavarait nagyobb gyakorisággal és nem ritkán súlyosabb formában észlelhetjük malignus megbetegedésekben [18]. Ez a diabetológiai és onkológiai klinikumban azt jelenti, hogy egyrészt a diabéteszesek közt gyakrabban szembesülünk különböző rákbetegségek kialakulásával, másrészt pedig - kissé bizarr módon - nem ritkán a malignus tumor megjelenése hívja fel a figyelmet a szénhidrátháztartás háttérben megbúvó, gyakran teljességgel tünetmentes zavaraira, látens prediabétesz vagy éppen manifeszt cukorbetegség fennállására. Ezen esetekben a korai stádiumú daganat a szénhidrát-anyagcsere zavarának egyik "első jeleként" is értékelhető "szimptóma".
A rosszindulatú daganatokhoz társult szekunder cukorbetegség alapvetően kétféle formában jelentkezhet, vagy a tumor által is generált inzulinrezisztencia talaján kialakult paraneopláziás kórformáról (T2DM-szerű altípus) vagy konkrét szervkárosodás, vagyis a hasnyálmirigy β-sejtjeinek immuncellulitisze következtében kialakult valódi pankreatogén diabéteszről (T1DM-szerű altípus) van szó. A rákbetegek onkoterápiája és obszervációja során viszonylag gyakran fordul elő az a helyzet is, amikor közvetlenül a tumorhoz társult vagy közvetve a hagyományos citosztatikumok (pl. fluorouracil, capecitabine stb.) és modern célzott biológiai terápiák (pl. sirolimus, everolimus, temsirolimus stb.), valamint az egyéb palliatív és szupportív kezelések (pl. szteroidok, szomatosztatin-analógok stb.) nemkívánatos hatásaként létrejött másodlagos diabétesszel találkozhatunk [19, 20, 21, 22]. Az onkoterápiák által okozott szénhidrátanyagcsere-zavarok súlyos formái klinikai megjelenésüket tekintve túlnyomó többségükben a szekunder cukorbetegség T2DM-szerű altípusába tartoznak, ám néhány újabb daganatellenes szer (pl. pembrolizumab, nivolumab stb.) kapcsán inkább a T1DM-szerű altípus kialakulása jellemző.
METABOLIKUS ÖSSZEFÜGGÉSEK
Hyperinsulinaemia, inzulinrezisztencia és tumorprogresszió
A daganatos betegek szervezetében kialakult inzulinrezisztens állapot, mint az inzulinrezisztencia egy sajátos, tumorhoz társult vagy paraneopláziás formája, az onkogenezis egyik fontos eleme. Ez részben a rákbetegséget megelőző, részben a tumoros szervezetben eleve meglévő metabolikus tényező, amely elsődlegesen a szénhidrátháztartás zavaraira kezdetben jellemző, kompenzációs vagy reaktív jellegű hyperinsulinaemia által okozott mitogén állapotnak felel meg. Kialakulására nagyban hajlamosít a magas glikémiás indexű szénhidrátokban és telített zsírokban gazdag, antioxidánsokban szegény étrend, a mozgáshiányos életmód, az elhízás, az alvászavar, a férfi nem, az idős kor, illetve bármilyen mértékű alkoholfogyasztás és dohányzás is [23]. A tumorhoz társult perifériás inzulinrezisztencia alapvetően azt jelenti, hogy - ellentétben az inzulinszenzitív daganattal - a gazdaszervezet bizonyos szöveteiben, elsősorban a zsír- és izomszövetekben, a normál mennyiségű inzulin nem képes elindítani vagy megfelelő szintre potencírozni az egyébként általa kiváltandó fiziológiás anyagcsere-változásokat. Ennek elsődleges oka a célsejtek alacsony válaszkészsége, ami főként az inzulinreceptorok - szerzett vagy öröklött genetikai mutációk által determinált - csökkent vagy hibás sejtfelszíni megjelenésével magyarázható.
Daganatos betegekben a ráksejtek által termelt számos citokin (pl. TNFα, IL-1, IL-2, IL-6, IL-8, IFN, PIF stb.) közvetlen vagy közvetett szerepet játszhat az inzulinreceptorok nem megfelelő expressziójában, az inzulin és az IGF-ek megváltozott hatékonyságában, s ezáltal indukál inzulinrezisztenciát [24, 25]. Ezt alátámasztó adat, hogy a TNF-gén overexpresszióját találták diabéteszesek vázizomzatában, ami eléggé egyértelmű bizonyítéka lehet a rosszindulatú daganat gazdaszervezetre gyakorolt, inzulinrezisztenciát előidéző direkt effektusának [26]. Tehát rákbetegség esetén maga a tumorszövet is előidézhet olyan, a gazdaszervezet szöveteit érintő változásokat, amelyek hosszabb-rövidebb távon - egyébként magának a tumornak kedvező, de a gazdaszervezet számára kedvezőtlen - egyfajta paraneopláziás inzulinrezisztencia kialakulásához, sőt akár a primer diabéteszhez nagyon hasonló, másodlagos cukorbetegség manifesztációjához vezethetnek [27].
Az inzulinrezisztencia korai fázisában létrejövő reaktív hyperinsulinaemia átmenetileg ugyan közel normoglykaemiás állapotot képes biztosítani a szervezetben, viszont ezzel egyidejűleg a tumorprogresszió kockázata is megnövekszik. Az inzulinreceptor bizonyos mutációi következtében ugyanis ilyenkor gyakran elvész az inzulin transzmembrán glükóztranszportot és glikogénszintézist serkentő hatása, viszont a mitogén, vagyis a sejtproliferációt elősegítő effektusa megmarad [28]. Jelenlegi tudásunk szerint az inzulin a közismert endokrin hatásain túl növekedési faktorként is funkcionál. Önmagában nem onkogén, vagyis közvetlenül nem okoz a tumorképződésben szerepet játszó génhibákat, s nem idézi elő a onkopatogén mutációkat nem tartalmazó, egészséges sejtek rákos elfajulását, viszont mitogén, azaz serkenti a már meglévő daganatsejtek növekedését és proliferációját [29]. Ezt a hatását nagyban fokozza, hogy a gazdaszervezetben, elsősorban a májszövetben, elősegíti az ún. inzulinszerű növekedési faktorok (IGF-ek), más néven szomatomedinek fiziológiás termelődését, továbbá erősíti azok saját mitogén aktivitását is [30]. A helyzetet bonyolítja, hogy általában magukban a daganatos sejtekben is patológiás jellegű IGF-termelés folyik. Az inzulin-, az IGF- és a vércukorszint együttes emelkedésének jelentős roboráló hatása van, amely a gazdaszervezet perifériás inzulinrezisztenciája miatt elsődlegesen az inzulinra relatíve érzékenyebb tumorszöveten érvényesül. Ezáltal a daganatsejtek a vérben keringő inzulint, IGF-eket és glükózt is - egyfajta "steal" effektus révén - a gazdaszervezettől magukhoz vonzzák. Következményesen fokozódik a sejtproliferáció és a klinikai tumorprogresszió, azaz növekszik a tumorgócok mérete és száma, illetve súlyosbodik az esetleges környezeti propagáció és a távoli disszemináció mértéke is. Mindez a teljes tumortömeg folyamatos növekedését vonja maga után, ami az említett "steal" jelenség automatikus felerősödésével jár. A gazdaszervezet állapota ezáltal is egyre gyorsabban sodródik a tumoros cachexia irányába, ez pedig egyfajta circulus vitiosusként tovább rontja a rákbetegek túlélési esélyeit [7].
Hyperglykaemia, nem-enzimatikus glikáció és tumorpromóció
A diabéteszes szénhidrátanyagcsere-zavar alapját képező inzulinrezisztencia kései szakaszára jellemző hyperglykaemia tumorpromóciót és tumorprogressziót elősegítő effektusa mintegy rárakódik a cukorbetegség korai stádiumainak metabolizmusát jellemző inzulin- és IGF-túlkínálat által okozott mitogén hatásokra. A tartósan elhúzódó hyperglykaemia fokozza azt a szöveti glikációt, amely egyébként az egészséges szervezetben is jelen van. Az ún. nem-enzimatikus glikáció során a glukóz kovalens kötéssel kapcsolódik nukleinsavak, struktúr- és enzimfehérjék, valamint lipidek szabad aminosav-csoportjaihoz, aminek eredményeként, több lépésben, az ún. Maillard-reakcióban, DNS-, fehérje- és lipid degradációt követően ún. fejlett glikációs végtermékek (AGE) képződnek [31, 32]. Ezen termékek egy része maga is intenzíven DNS-, sejt- és szövetkárosító anyag (pl. enediol, dialdehidek, imidazolon, pentozidin stb.), más része pedig további, még erőteljesebben toxikus ágensek, reaktív oxigén-származékok (pl. szuper-oxid, hidrogén-peroxid stb.) és egyéb szabadgyökök képzésében vesz részt. Fiziológiás mértékű glikáció során a keletkező destruktív szabadgyökök eliminációja egyensúlyt tart a képződésükkel. Patológiás esetben viszont - így manifeszt cukorbetegségben, sőt már prediabéteszben is - ez az egyensúly felborul, s a glikációs változások a DNS-, sejt- és szövetkárosodásoknak az oldalára tolódnak el. Mindennek a szervezetre gyakorolt legfontosabb negatív következménye egy elhúzódó és kontrollálatlan oxidatív stressz állapot kialakulása, amiben a toxikus ROS és a sejtvédő endogén antioxidáns vegyületek (pl. glutation stb.) közt tartós egyensúlyhiány jön létre, amely akár évekig is elhúzódhat [33]. Az egyensúly felborulása a fiziológiás öregedési folyamatok és az atherosclerosis felgyorsítása mellett idővel számos krónikus betegség, így a malignus daganatos kórképek kialakulásához is elvezethet [34].
A metabolizmus minden területét érintő glikációs változások eredményeként az oxidatív stressz állapotába került szervezetben a glikáció direkt DNS-károsító hatásán túl a genom instabilitását indirekt módon előidéző effektusáról sem szabad megfeledkezni, amelyért epigenetikai változások sora, így a hisztonfehérjék, a hiszton-acetilázok és -deacetilázok, a DNS-metilázok és -demetilázok, a DNS-metiltranszferázok, valamint a különféle speciális DNS repair enzimek kémiai módosítása felelős. A szabadgyökök szövetkárosító hatását a képződésük megelőzésére irányuló egészséges életmóddal, megfelelő táplálkozással és rendszeres testmozgással, valamint természetes és mesterséges antioxidánsok direkt alkalmazásával jelentősen mérsékelhetjük. Emellett nem szabad elfeledkeznünk arról sem, hogy ezt a fajta - toxikus szabadgyökök által előidézett - szövetdestrukciós effektust akár gyógyító célra is felhasználhatjuk. A daganatellenes indikációkban alkalmazott radioterápia rákszövetet pusztító hatása, többek között, ezen az elven alapul.
Mindezek után megállapíthatjuk, hogy a diabéteszre általában jellemző fokozott tumorkockázat - metabolikus szempontból - elsődlegesen az elhúzódó hyperglykaemia bonyolult biokémiai hatásaira és a következményes nem-enzimatikus glikációs remodellingre vezethető vissza, amely egyaránt segíti a kóros sejtek képződését és terjedését, valamint a sejtnövekedést és a sejtproliferációt is [5]. A glikációs változások a karcinogenezis többlépcsős modelljében elvileg minden szinten jelen lehetnek, viszont feltételezhetően önmagukban általában nem elégségesek a rákképződési kaszkád teljes végbemeneteléhez. Alapvetően a tumorpromócióban és a tumorprogresszióban játszhatnak jelentősebb szerepet, míg a tumoriniciációért, vagyis a daganatképződés első lépcsőjét jelentő DNS-mutáció létrejöttéért inkább különféle erélyes fizikai vagy kémiai karcinogén ágensek tehetők felelőssé [35]. Ezen gondolatmenet alapján érthető módon a 2-es típusú diabétesz a már kialakult rákbetegség progresszióját vélhetően azáltal is elősegíti, hogy aktiválhatja a szervezetben rendszeresen és rövid időre megjelenő, majd eltűnő, vagy esetleg a G0-fázisban mégis tartósan megmaradt ún. "alvó" ráksejteket [36].
MOLEKULÁRIS ÖSSZEFÜGGÉSEK
IGF-ek szerepe és hatásmechanizmusa
Az inzulin- és IGF1-receptorokat az inzulin, az IGF1 és az IGF2 egyaránt aktiválhatja, s hatásukra működésbe lép a RAS/RAF/MEK/ERK/MAPK, röviden RAS és a PI3K/AKT/mTOR, röviden PI3K jelátviteli útvonal is [37, 38]. Az IGF2-receptorok IGF2-kötése ugyanakkor az említett jelátviteli utakra egy fentiekkel ellentétes, szuppresszor hatást fejt ki. Az IGF-ek aktivitását a hozzájuk nagy affinitással kapcsolódó IGF-kötő fehérjék (IGFBP-k) általában azáltal csökkentik, hogy a kisebb affinitású IGF1R hozzáférését az IGF1 és IGF2 molekulákhoz akadályozzák. Számos sejtproliferációt gátló, apoptózist segítő, igazolt vagy feltételezett tumorellenes effektusáról ismert fehérje (pl. p16, p53, PTEN, TSC stb.), hormon (pl. antiösztrogén stb.), vitamin (pl. retinoid, D-vitamin stb.) és egyéb anyag (pl. TGF-β stb.) is azáltal csökkenti az IGF-ek hatását, hogy fokozzák az IGFBP-k termelését. Megjegyzendő, hogy bizonyos esetekben éppenséggel egyes, IGF-eket kisebb affinitással kötő IGFBP-altípusok fokozott termelése ronthatja a tumorprognózist [39]. Az IGF-ek az általuk aktivált intracelluláris jelátviteli rendszeren keresztül antiapoptotikus, proliferatív és differenciálódást elősegítő hatást váltanak ki. A vérben fiziológiásan elérhető mennyiségüket az IGFBP-k szabályozzák. A receptor-ligand kapcsolódást követően létrejövő tirozin-kináz-aktivitás két fő irányban, egyrészt a proliferatív szignált közvetítő RAS-útvonalon, másrészt az antiapoptotikus jeleket továbbító PI3K-útvonalon keresztül fejti ki hatását a sejtciklusra. E két útvonal és a hozzájuk csatlakozó egyéb szignálutak között számos kapcsolat van, de az egyik legnagyobb jelentőségű mégis talán az, hogy az aktivált RAS képes önállóan is beindítani a PI3K jelutat. Ezáltal ugyanis az egyszerre érvényesülő proliferatív és apoptózisgátló jelek egymást gyakorlatilag szinergista módon felerősítve robbanásszerű sejtburjánzást idézhetnek elő [40].
Fontos molekuláris összefüggés a 2-es típusú diabétesz és a daganatok kialakulása közt az, hogy az IGF1R-aktiváció a PI3K-útvonalon keresztül fokozza az intracitoplazmatikus vezikulákban raktározott GLUT1 transzmembrán fehérje sejtfelszíni expresszióját, ami elősegíti a glükóz passzív transzportját és felvételét azokba a ráksejtekbe, amelyeknek a glükóz-igénye a metabolizmusukat gyakran jellemző aerob glikolízis, az ún. Warburg-effektus miatt egyébként is jelentősen megnövekedett. Számos tumor esetén az IGF1R vagy a GLUT1 extrém mértékben fokozott sejtfelszíni expressziójához vezető génmutációk kialakulása jelenti a többlépcsős tumorgenezis egyik lépcsőfokát (pl. gyomorrák, GIST, osteosarcoma, emlőrák, myeloma multiplex, Hodgkin-lymphoma, kissejtes tüdőrák esetén) [41]. Mindebből kiindulva korábbi gyógyszerkutatások során logikus gondolatként merült fel az, hogy IGF-receptorok daganatellenes terápiák targetjeként szerepelhetnek, azonban még számos molekuláris összefüggést ma sem ismerünk e téren. Erre az egyik legjobb bizonyíték az, hogy az eddig kifejlesztett célzott anti-IGFR monoklonális antitest (MAB) vagy IGFR-tirozin-kináz-inhibitor (TKI) terápiák legnagyobb része a klinikai vizsgálatok II-III. fázisában jelentős mértékű toxicitás, esetleg elégtelen vagy paradox tumorválasz miatt visszavonásra került (pl. cixutumumab, figitumumab, dalotuzumab, robatumumab, ganitumab, linsitinib stb.), s csupán alig néhányukat sikerült daganatellenes vagy más indikációban törzskönyvezni (pl. elotuzumab stb.) [39].
A RAS- és PI3K-útvonalakról általában
Ma már biztosan tudjuk, hogy az egyik legősibbnek tartott RAS-MAPK és a vele számtalan módon kötődő PI3K-mTOR jelutak a cukorbetegség és a rákbetegség kialakulásában egyaránt központi jelentőséggel bírnak. Részleteik és kapcsolódási pontjaik alaposabb megismerése bizonyosan közelebb vihet a látszólag független kétféle kórkép összefüggéseinek mélyebb megértéséhez. A T2DM kialakulásának és fennállásának különböző szakaszaiban az inzulin és a szomatomedinek (IGF1 és IGF2) eltérő intenzitással és affinitással képződnek, így változó mértékben és módon stimulálják az inzulin- és IGF-receptorokhoz intracellulárisan csatlakozó RAS és PI3K utakat, amelyek ezután a sejtplazmában és a sejtmagban is számos folyamat szabályozásában vesznek részt. Befolyásolják a sejtek növekedését, proliferációját és differenciálódását, az apoptózist (=programozott sejthalál) és az autofágiát (=önemésztés), az angiogenezist, a sejtek energia-, oxigén- és tápanyagellátását, fehérje- és lipidszintézisét, valamint az inflammációs és stressz reakciókat, vagyis összességében a sejtek túlélését. A sejtnövekedés és -proliferáció fokozása, illetve a differenciáció és az apoptózis gátlása, valamint az autofágia se nem alacsony, se nem túlzott mértékű fenntartása a rosszindulatú daganatok képződésének szinte obligát elemei. Tehát az apoptózisnak és az autofágiának a glükózmetabolizmus és az energiatermelés általi megfelelő kontrollálása (cell surveillance) alapvető fontosságú a sejtek homeosztázisának és túlélésének biztosításában [42].
Az inzulin és a szomatomedinek metabolikus hatásainak fő közvetítője a PI3K-út. Fokozza a glikogén- és fehérjeszintézist, valamint a sejtek glukózfelvételét. Glükóz jelenlétében az AMPK gátlása közvetve aktiválja a PI3K-útvonal végén lévő mTORC1-et, ami viszont gátolja az autofágiát. Az energiaszint csökkenése, vagyis alacsonyabb ATP-szint és magasabb AMP-szint mellett az AMPK aktiválódik, ami viszont az mTORC1 komplexet foszforilálja és inaktiválja, ez pedig fokozza az autofágiát. A túlzott autofágia végül a sejt pusztulásához vezethet, amit autoapoptózisnak is nevezünk [43]. Mindez azt jelenti, hogy az AMPK aktiválása jelentős részben hat a daganatképződés ellen. Itt említhető meg, hogy a metformin direkt tumorellenes hatása is elsődlegesen az AMPK aktiválásán keresztül érvényesül. Az ezzel ellentétesen ható, vagyis az AMPK inaktiválását kiváltó tényezők ugyanakkor az onkogenezis támogatói. Az LKB1 tumorszuppresszor gén terméke egy olyan szerin/treonin-kináz, amely az AMPK aktiválása révén köti össze a tápanyag- és energiafelhasználásnak, valamint a sejtszerkezet, a sejtváz (citoszkeleton) reorganizációjának, ezzel együtt pedig a sejtek folyamatos megújulásának, vagyis túlélésének irányítását. Ebből következően megérthető, hogy az LKB1 gén recesszív, biallélikus, gátló jellegű mutációi számos daganatos betegség (pl. CRC, tüdő-, méhnyak-, emlőrák stb.) létrejöttében és progressziójában is patológiás szerepet játszhatnak [44]. Mindezen ismeretek alapján készült saját sémás rajzunkkal az alábbiakban elsősorban azt próbáljuk érzékeltetni, hogy a diabétesz és a rák viszonyának vonatkozásában központi jelentőséggel bíró RAS- és PI3K-útvonalak a rendkívül szerteágazó molekuláris kapcsolataik révén a sejtműködés szinte minden részét elérik, s azokat változó módon befolyásolhatják is [1. ábra].
1. ábra | A RAS- és PI3K-útvonalak viszonya. Molekuláris kapcsolódási pontok a malignus daganat és a T2DM kialakulásában (saját ábra)
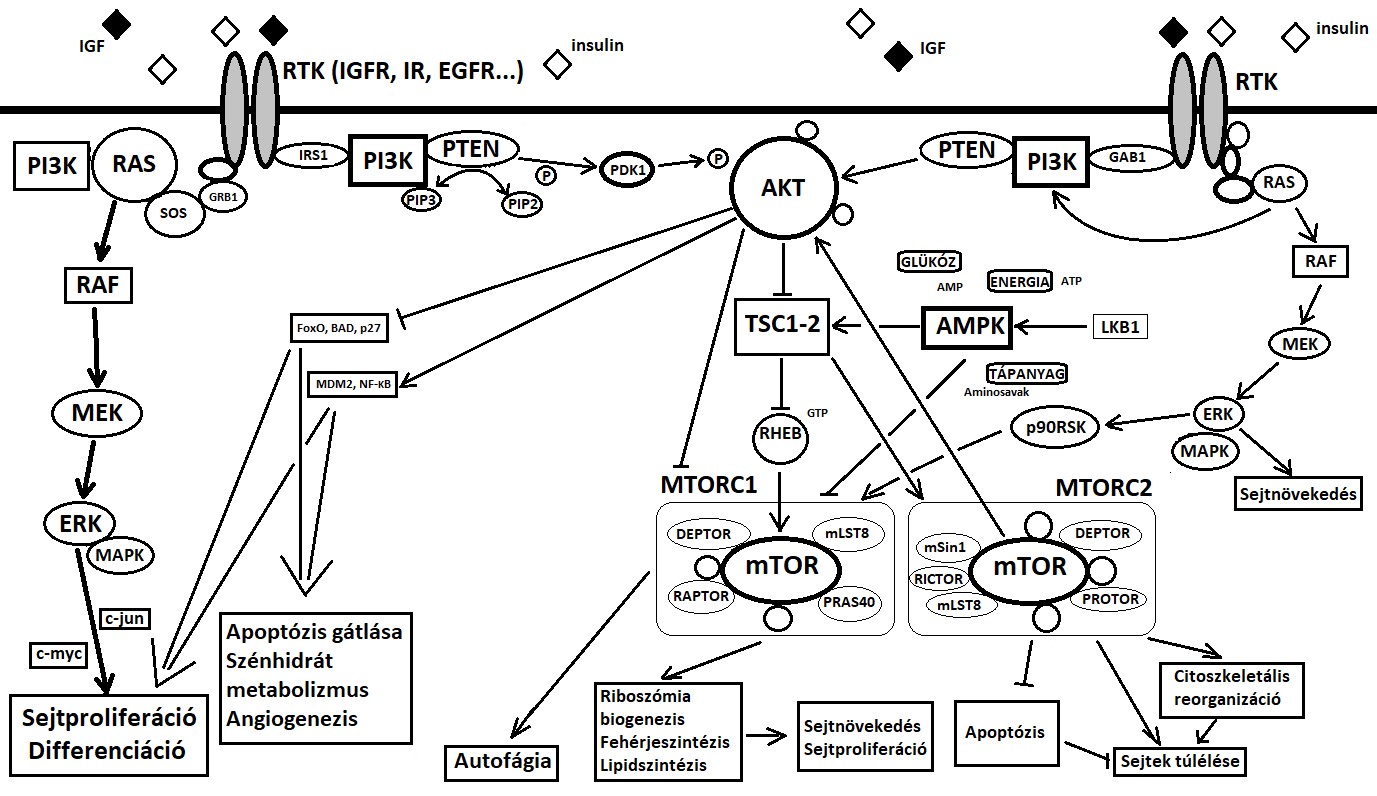
Deptor: (DEP domain containing mTOR-interacting protein); GTP: (guanosine triphosphate) guanozin-trifoszfát; mLST8: (mammalian lethal with SEC13 protein 8); mSin1: (mammalian stress-activated protein kinase-interacting protein 1); p90RSK: (p90 ribosomal S6 kinase) p90 riboszómális S6 kináz; PDK: (pyruvate dehydrogenase kinase) piruvát-dehidrogenáz kináz; PIP: (phosphatidylinositol phosphate) foszfatidil-inozitol-foszfát; PRAS40: (proline-rich Akt substrate 40 kDa) prolin-gazdag Akt szubsztrát 40 kDa; Protor: (protein observed with Rictor); Raptor: (regulatory associated protein of mTOR) mTOR szabályozásához kapcsolódó fehérje; RHEB: (RAS homolog enriched in the brain); Rictor: (rapamycin-insensitive companion of mTOR); SOS: (son of sevenless)
(Az ábrában és a közlemény szövegében is szereplő rövidítéseket lásd a közlemény előtti Rövidítések jegyzékében!)
Jelátviteli utak szerepe a diabéteszesek fokozott rákkockázatában
Prediabétesz vagy manifeszt cukorbetegség esetén a képződő növekedési faktorok (pl. inzulin, IGF, EGF, VEGF stb.) a tumorsejtek megfelelő tirozin-kináz receptoraival (RTK) szemben fokozott affinitást mutatnak. Az általuk aktivált receptorokhoz ún. dokkoló fehérjék (pl. IRS1-2, GAB1 stb.) csatlakoznak, amelyek - egyfajta adapterként - különböző módokon, akár egyidejűleg is, többféle jelátviteli út beindítására képesek. Az egyik ilyen út a mitogén szignált továbbító RAS-útvonal, amelynek végén lévő aktivált MAPK/ERK komplexek a sejtmagba jutva transzkripciós faktorokat stimulálnak. A RAS-útvonal aktivációját tehát a MAPK/ERK komplexek közvetítik a nukleáris transzkripciós faktorok felé (pl. c-Myc, c-Jun, c-Fos, Mad, Max, Elk-1, p53 stb.), melyek további aktivációja serkenti a sejtproliferációt. Közismert tény, hogy ezek a faktorok általában olyan gének, ún. protoonkogének termékei, amelyek domináns, monoallélikus, aktiváló jellegű mutációi, amplifikációi elmaradhatatlan lépései lehetnek számos rosszindulatú daganat többlépcsős tumorgenezisének. A c-myc gén terméke az összes többi gén 15%-ának expresszióját szabályozza, így érthető, hogy ezen transzkripciós fehérje (c-Myc) túlzott termelése számos daganat képződésében központi szerepet játszhat (pl. Burkitt-lymphoma, myeloma, melanoma, CRC, NSCLC, emlő- és prosztatarák esetén). A helyzetet tovább komplikálja, hogy a MAPK többféle módon is képes szabályozni azokat a transzkripciós faktorokat, amelyek révén érvényesül a MAPK-aktiváció mitogén effektusa növekedési faktorok (pl. IGF, EGF, FGF, VEGF, PDGF stb.), anabolikus hormonok (pl. inzulin, pajzsmirigyhormonok, ösztrogének stb.) vagy éppen thrombin hatására [45]. A YAP a sejtproliferációt és az apoptózist is reguláló Hippo-jelátviteli útvonal downstream effektora. A YAP-fehérje képződését a c-myc és c-jun gének expressziója is fokozhatja, ami esetenként fontos szereppel bírhat az onkogenezisben (pl. HCC, hasnyálmirigyrák stb.). Ide kapcsolódó adat, hogy a c-jun gén mutációi egyébként relatíve gyakran fordulnak elő emlő-, prosztata- és tüdőrákban [46].
A RAS-útvonal tagjainak aktiváló mutációi számos tumorban kimutathatók és célzott biológiai terápiák kiemelt targetjeit jelentik (pl. tüdő-, húgyhólyag-, hasnyálmirigy-, pajzsmirigyrák, RCC, CRC, HCC, melanoma, seminoma és AML esetén). A RAS jelutat bénító terápiák közül a gyakorlatban szélesebb körben egyelőre a RAF- és a MEK-mutációkra ható gátló szerek terjedtek el, míg a direkt RAS-inhibitorok (pl. tipifarnib, lonafarnib stb.) inkább csak klinikai vizsgálatokban és egyes ritka betegségek ellátásában szerepelnek. A mindennapi onkológiai betegellátásban használt RAF-gátlók közül legismertebbek a vemurafenib, dabrafenib és encorafenib, illetve a MEK-inhibitorok csoportjában pedig a trametinib, cobimetinib és binimetinib, melyek mindegyikét eredetileg melanoma malignum kezelésére törzskönyvezték, ma viszont már egyre több - BRAF V600 mutációt hordozó - egyéb tumor (pl. NSCLC, CRC stb.) kezelésében is felmerül terápiás indikációjuk [47, 48].
A daganatképződésben igen gyakran szerepet játszó aktiváló jellegű PI3K-mutációk és amplifikációk (pl. PIK3CA, PIK3R1 mutáció stb.) a szignalizációt folyamatosan fenntartják és "gyorsítják" (pl. fej-nyaki, nyelőcső-, gyomor-, emlő-, petefészek-, endometrium- és méhnyakrák, tüdő laphámrákja, CRC, melanoma, glioblastoma esetén), ugyanakkor a normál (vad típusú) PTEN és TSC 1-2 tumorszuppresszor gének termékei pedig bénítják vagy "lassítják" a PI3K jelutat. Az utóbbi géneket érintő gátló jellegű mutációk szintén szerepet játszathatnak a kontrollálatlan tumorsejt-profileráció létrejöttében (pl. RCC, húgyhólyag-, prosztata-, emlő-, petefészek- és endometriumrák, melanoma, glioblastoma esetén). A PI3K-útvonal egyik központi eleme a szerin/treonin-kináz aktivitású AKT enzimcsalád, amelynek három tagja van. Mindegyikük számos proapoptotikus, vagyis apoptózist elősegítő, kináz aktivitású enzimet gátol (pl. p21, p27, FoxO, BAD stb.), ugyanakkor többféle antiapoptotikus transzkripciós faktort (pl. NFκB, MDM2 stb.) pedig aktivál. Ezeknek köszönhetően az aktivált AKT a sejtben gyakorlatilag az egyik legerősebb apoptotózisgátló szignál, amelynek aktiváló jellegű mutációi és amplifikációi számos daganatos betegségben fellelhetőek. Az AKT1 mutációja CRC esetén, emlő-, petefészekrákban és a tüdő laphámrákjában, amplifikációja gyomorrákban, az AKT2 mutációja CRC esetén, amplifikációja emlő-, petefészek-, hasnyálmirigyrákban és a fej-nyak laphámrákjában, az AKT3 mutációja melanomában, amplifikációja pedig emlőrákban gyakori [49].
A PI3K-útvonal utolsó tagja az mTOR kináz, amely a sejtben két komplexben található: az mTORC1 és az mTORC2 formájában. Az mTORC1 a sejtciklus szabályozásában központi szereppel bír, számos irányból - így akár a RAS-útvonalról - érkező jeleket is képes fogadni és transzformálni. Jellemzően inzulin és IGF, illetve egyéb anabolikus hormonok és növekedési faktorok hatására fokozza a fehérjeszintézist és a sejtproliferációt. Az mTORC2 a sejtek túlélését, a citoszkeleton reorganizációját modulálja. Mindezek alapján érthető, hogy a PI3K-útvonalat létrehozó enzimfehérjék génjeiben (pl. PIK3CA, PIK3R1, PTEN, TSC1-2 stb.) bekövetkező, aktiváló vagy gátló jellegű mutációk és amplifikációk közvetve vagy közvetlenül az mTOR enzim serkentése révén tumorpromóciót elősegítő szerepet játszhatnak. Ezt támasztja alá az a gyakorlati tény is, hogy a PI3K-útvonal végén lévő mTORC1 és 2 komplexek gátlása igen erőteljes tumorellenes hatással bír. A napjainkban használatos mTOR-inhibitor szerek közül a temsirolimus RCC és köpenysejtes lymphoma, míg az everolimus RCC, NET és emlőrák kezelésében rendelkezik törzskönyvi indikációval [50].
ÖSSZEFOGLALÁS ÉS KÖVETKEZTETÉS
A szénhidrát-anyagcsere zavarai és a rosszindulatú daganatok közötti összefüggést számos preklinikai és klinikai vizsgálat, valamint nagy metaanalízisek eredményei is alátámasztják [1, 2, 8]. A kétféle kórállapot közötti szoros kapcsolat okát egyrészt a hasonló anyagcsere-változások, másrészt a közös molekuláris utak jelentik. A 2-es típusú cukorbetegségre jellemző megnövekedett rákkockázat leginkább az glükóz-metabolizmus diabéteszt megelőző és kísérő változásainak, a szisztémás inzulinrezisztenciának, a reaktív hyperinsulinaemiának és az elhúzódó hyperglykaemiának lehet a következménye. Az inzulinrezisztencia és a hyperinsulinaemia inkább a tumorprogressziót fokozza, míg a hyperglykaemia - a többlépcsős karcinogenezis minden szintjén előidézett nem-enzimatikus glikációs változások révén - a tumorpromócióra is jelentős serkentő hatást gyakorol. Bár a tumoriniciációban a glikációs folyamatok során képződő toxikus szabadgyökök oki szerepe erősen valószínűsíthető, azonban ennek egyértelmű bizonyításához még további alapkutatásokra van szükség.
A tumorszövet - ellentétben az inzulinrezisztenssé váló gazdaszervezettel - inzulinra és egyéb növekedési faktorokra fokozottan érzékenyen reagál. Mindez többféle mechanizmus révén segíti a daganat növekedését, illetve fokozza a gazdaszervezet idült senyvedését, a cachexia kialakulását. Az inzulin- és IGF-receptorok aktiválásán keresztül a tumorban számos intracelluláris jelút kapcsolódik be vagy bénul meg a felgyorsult intratumorális anyagcsere által előidézett megnövekedett energiaigény miatt előálló relatív energiahiány és a környezetből érkező időszakosan fokozott glükóztúlkínálat miatt. Ezen folyamatok szabályozása a RAS és PI3K jelátviteli útvonalak molekuláris összefüggéseinek teljes körű feltárása nélkül nem érthetők meg. A RAS-RAF-MEK-ERK-MAPK útvonal a sejtproliferáció, a PI3K-(PTEN)-AKT-(TSC)-mTOR útvonal pedig az apoptózisgátlás szignálját továbbítja a megfelelő sejtalkotórészek felé. A két jelút egyidejű aktivációja és a jelutak alkotásában részt vevő egyes enzimfehérjék bizonyos mutációi egymás hatását felerősítve a daganatsejtek rapid felszaporodásához vezethetnek. A RAS- és a PI3K-útvonalakat érintő szerzett génhibák fontos targetet jelentenek a célzott biológiai terápiák kifejlesztésében. Ma már többféle malignus daganat kapcsán számos RAS-, RAF-, MEK-, ERK-, MAPK-, PI3K-, AKT- és mTOR-inhibitorral zajlanak klinikai vizsgálatok. Közülük többet már évekkel ezelőtt törzskönyveztek, illetve jelenleg is a klinikai gyakorlatban alkalmazunk.
Végezetül tehát kijelenthetjük, hogy a T2DM és a rákbetegség összefüggéseinek hátterében álló, rendkívül komplex molekuláris kapcsolatok további kutatása, részletes feltárása központi jelentőségű a közös patomechanizmusok és terápiás támadáspontok felismerésében. Az antidiabetikumok tekintetében ez a kutatómunka nagyban segítheti a tumorellenes vagy tumorkockázat-növelő effektusok kiaknázását vagy megelőzését, illetve a citosztatikumok tekintetében pedig az esetleges diabetológiai mellékhatások megértését és kivédését. A kétféle betegség molekuláris viszonyainak teljes feltérképezése, a jövőbeni gyógyszerfejlesztések várható jelentős költségei, valamint a jelenleg is könnyen hozzáférhető, nagyszámú, kiváló hatékonyságú és megfelelően biztonságos antidiabetikum miatt, reálisan csak hosszú évek múlva eredményezheti a célzott onkológiai kezelésekhez hasonló, precíziós jellegű antidiabetikus terápiák bevezetését. Az viszont már ma is gyakorlati pozitívum, hogy a diabéteszes vagy akár a nem-diabéteszes rákbetegek onkoterápiájában egyes esetekben "személyre szabott" modern antidiabetikum-antineoplasztikum kombinációk alkalmazására is lehetőségünk nyílik a maximális gyógyulási esély érdekében.
IRODALOM
1. Pearson-Stuttard J, Papadimitriou N, Markozannes G, et al. Type 2 diabetes and cancer: an umbrella review of observational and mendelian randomization studies. Cancer Epidemiol Biomarkers Prev. 2021; 30: 1218-1228.
2. Ling S, Brown K, Miksza JK, et al. Association of type 2 diabetes with cancer: a meta-analysis with bias analysis for unmeasured confounding in 151 cohorts comprising 32 million people. Diabetes Care. 2020; 43: 2313-2322.
3. Zhu B, Qu S. The relationship between diabetes mellitus and cancers and cancers and its underlying mechanisms. Front Endocrinol. 2022; 13: 800995.
4. Brown JC, Carson TL, Thompson HJ, et al. The triple health threat of diabetes, obesity, and cancer-epidemiology, disparities, mechanisms, and interventions. Obesity. 2021; 29: 954-959.
5. Ryu TY, Park J, Scherer PE. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab J. 2014; 38: 330-336.
6. Poznyak A, Grechko AV, Poggio P, et al. The diabetes mellitus-atherosclerosis connection: the role of lipid and glucose metabolism and chronic inflammation. Int. J. Mol. Sci. 2020; 21: 1835.
7. Bánhegyi RJ, Rus-Gal PO, Nagy ÁK, et al. Antidiabetic therapy - a new possibility in the complex therapy of cancer? [Antidiabetikus kezelés, mint újabb lehetőség a daganatok komplex terápiájában?] Magy Onkol. 2010; 54: 315-323. [Hungarian]
8. Yang K, Liu Z, Thong MSY, et al. Higher incidence of diabetes in cancer patients compared to cancer-free population controls: a systematic review and meta-analysis. Cancers. 2022; 14: 1808.
9. Qi L, Qi X, Xiong H, et al. Type 2 diabetes mellitus and risk of malignant melanoma: a systematic review and meta-analysis of cohort studies. Iran J Public Health. 2014; 43: 857-866.
10. Wang Y, Liu X, Yan P, et al. Association between type 1 and type 2 diabetes and risk of non-Hodgkin's lymphoma: a meta-analysis of cohort studies. Diabetes Metab. 2020; 46: 8-19.
11. Li X, Wang X, Gao P. Diabetes mellitus and risk of hepatocellular carcinoma. Biomed Res Int. 2017; 2017: 5202684.
12. Mills KT, Bellows CF, Hoffman AE, et al. Diabetes mellitus and colorectal cancer prognosis: a meta-analysis. Dis Colon Rectum. 2013; 56: 1304-1319.
13. Petrov MS. Post-pancreatitis diabetes mellitus and excess intrapancreatic fat deposition as harbingers of pancreatic cancer. World J Gastroenterol. 2021; 27: 1936-1942.
14. Tseng C-H. Type 2 diabetes mellitus and kidney cancer risk: a retrospective cohort analysis of the National Health Insurance. PLoS One. 2015; 10(11): e0142480.
15. Zhu Z, Wang X, Shen Z, et al. Risk of bladder cancer in patients with diabetes mellitus: an updated meta-analysis of 36 observational studies. BMC Cancer. 2013; 13: 310.
16. Zhao X-B, Ren G-S. Diabetes mellitus and prognosis in women with breast cancer: a systematic review and meta-analysis. Medicine. 2016; 95: e5602.
17. Zhang Z-H, Su P-Y, Hao J-H, et al. The role of preexisting diabetes mellitus on incidence and mortality of endometrial cancer: a meta-analysis of prospective cohort studies. Int J Gynecol Cancer. 2013; 23: 294-303.
18. Vigneri P, Frasca F, Sciacca L, et al. Diabetes and cancer. Endocr Relat Cancer. 2009; 16: 1103-1123.
19. Feng J-P, Yuan X-L, Li M, et al. Secondary diabetes associated with 5-fluorouracil-based chemotherapy regimens in non-diabetic patients with colorectal cancer: results from a sinle-centre cohort study. Colorectal Dis. 2013; 15: 27-33.
20. Yim C, Hussein N, Arnason T. Capecitabine-induced hyperosmolar hyperglycaemic state. BMJ Case Rep. 2021; 14: e241109.
21. Goldman JW, Mendenhall MA, Rettinger SR. Hyperglycemia associated with targeted oncologic treatment: mechanisms and management. Oncologist. 2016; 21: 1326-1336.
22. Oyer DS, Shah A, Bettenhausen S. How to manage steroid diabetes in the patient with cancer. J Support Oncol. 2006; 4: 479-483.
23. Káplár M, Paragh Gy. Oral antidiabetics and cancer risk. [Orális antidiabetikumok és daganatkockázat.] Diabetol Hung. 2013; 21: 101-109. [Hungarian]
24. Wigmore SJ, Fearon KCH, Sangster K, et al. Cytokine regulation of constitutive production of interleukin-8 and -6 by human pancreatic cancer cell lines and serum cytokine concentrations in patients with pancreatic cancer. Int J Oncol. 2002; 21: 881-886.
25. Wigmore SJ, Fearon KCH, Maingay JP, et al. Effect of interleukin-2 on peripherial bood mononuclear cell cytokine production and the hepatic acute phase protein response. Clin Immunol. 2002; 104: 174-182.
26. Saghizadeh M, Ong JM, Garvey WT, et al. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J Clin Invest. 1996; 97: 1111-1116.
27. Arcidiacono B, Iiritano S, Nocera A, et al. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res. 2012; 2012: 789174.
28. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014; 6: a009191.
29. Smith U, Gale EAM. Does diabetes therapy influence the risk of cancer? Diabetologia. 2009; 52: 1699-1708.
30. Draznin B. Mitogenic action of insulin: friend, foe or 'frenemy'? Diabetologia. 2010; 53: 229-233.
31. Zeng C, Li Y, Ma J, et al. Clinical/translational aspects of advanced glycation end-products. Trends Endocrinol Metab. 2019; 30: 959-973.
32. Kuzan A. Toxicity of advanced glycation end products (Review). Biomed Rep. 2021; 14: 46.
33. Dabrowski M, Szymanska-Garbacz E, Miszczyszyn Z, et al. Risk factors for cancer development in type 2 diabetes: a retrospective case-control study. BMC Cancer. 2016; 16: 785.
34. Franceschi C, Garagnani P, Morsiani C, et al. The continuum of aging and age-related diseases: common mechanisms but different rates. Front Med. 2018; 5: 61.
35. Schwartz SS, Grant SFA, Herman ME. Intersections and clinical translations of diabetes mellitus with cancer promotion, progression and prognosis. Postgrad Med. 2019; 131: 597-606.
36. Sciacca L, Vigneri R, Tumminia A, et al. Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis. 2013; 23: 808-815.
37. Hua H, Kong Q, Yin J, et al. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy. J Hematol Oncol. 2020; 13: 64.
38. Vasas P, Winslet MC, Shi YY. The role of insulin-like growth factors (IGF) in cell division processes and in malignancy. [Az inzulinszerű növekedési faktor (IGF) szerepe a sejtosztódási folyamatokban és a daganatokban.] Orv Hetil. 2009; 150: 2308-2312. [Hungarian]
39. Kopper L, Tímár J, Becságh P, et al. Targeted diagnosis and targeted therapy in oncology 4. [Célzott diagnosztika és célzott terápia az onkológiában 4.] Semmelweis Kiadó, Budapest, 2015; page 100, 183. [Hungarian]
40. Wu P-K, Becker A, Park J-I. Growth inhibitory signaling of the Raf/MEK/ERK pathway. Int J Mol Sci. 2020; 21: 5436.
41. Sebestyén A, Hujber Z, Jeney A, et al. Tumormetabolism. [Tumormetabolizmus.] Klin Onkol. 2016; 3: 51-58. [Hungarian]
42. Yun CW, Lee SH. The roles of autophagy in cancer. Int J Mol Sci. 2018; 19: 3466.
43. Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017; 36: 1811-1836.
44. Korsse SE, Peppelenbosch MP, Veelen W. Targeting LKB1 signaling in cancer. Biochim Biophys Acta. 2013; 1835: 194-210.
45. Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020; 21: 2346.
46. Xiao W, Wang J, Ou C, et al. Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem Biophys Res Commun. 2013; 439: 167-172.
47. Dumaz N, Lebbé C. New perspectives on targeting RAF, MEK and ERK in melanoma. Curr Opin Oncol. 2021; 33: 120-126.
48. Malapelle U, Passiglia F, Cremolini C, et al. RAS as a positive predictive biomarker: focus on lung and colorectal cancer patients. Eur J Cancer. 2021; 146: 74-83.
49. Hua H, Zhang H, Chen J, et al. Targeting Akt in cancer for precision therapy. J Hematol Oncol. 2021; 14: 128.
50. Qiu H-Y, Wang P-F, Zhang M. A patent review of mTOR inhibitors for cancer therapy (2011-2020). Expert Opin Ther Pat. 2021; 31: 965-975.
LINK (Orvosi Hetilaphoz):
https://pubmed.ncbi.nlm.nih.gov/36153724/
















